
Documentation
JSpecies Documentation
JSpecies is an easy to use, biologist-centric software designed to measure the probability if two genomes belonging to the same species or not.
1. Starting JSpecies
JSpecies can be started via Webstart here or
alternative a .jar can be downloaded here.
Check System Requirements.
Information: If you download JSpecies you can start the .jar by typing:
java -jar -Xms512m -Xmx512m JSpecies.jar
2. Adjusting Preferences
If you start JSpecies for the first time you have to adjust the general parameters to your computer system. Open the Preferences Dialog through the menu item Edit. Choose here your workspace (all JSpecies data will be stored there), and point BLAST, NUCmer, formatDB and fastacmd to the executables.
Information: On Windows the executable are named blastall.exe, formatdb.exe and fastacmd.exe. They are located in your BLAST installation directory under:'\yourpath\bin\'
Read more about BLAST installation on Windows here. NUCmer is NOT available for Windows operating systems.
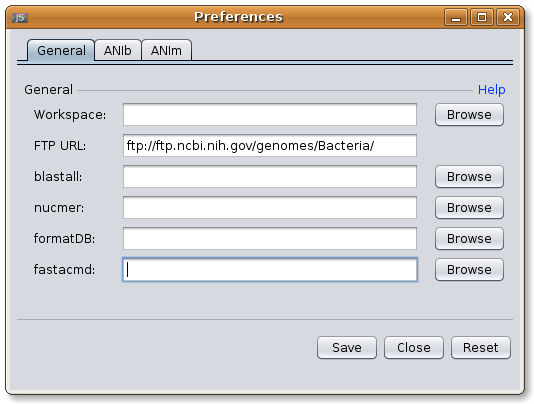 Image: JSpecies General Preferences Dialog.
Image: JSpecies General Preferences Dialog.ANIb parameters: JSpecies provides by default, evaluated parameters for the calculation of ANIb values (Goris et al., 2007). This parameters can be adjusted if needed. Implemented parameters are:
- -X = dropoff value for gapped alignment [Integer]
- -q = penalty for a nucleotide mismatch [Integer]
- -F = filter query sequence [T/F]
- -e = expectation value (E) [Real]
- -a = number of processors to use [Integer]
Warning: A JSpecies group can only be based on one set of parameters. Adjusting parameters for a group which already contains results will lead to the deletion of all results for ANIb calculations!
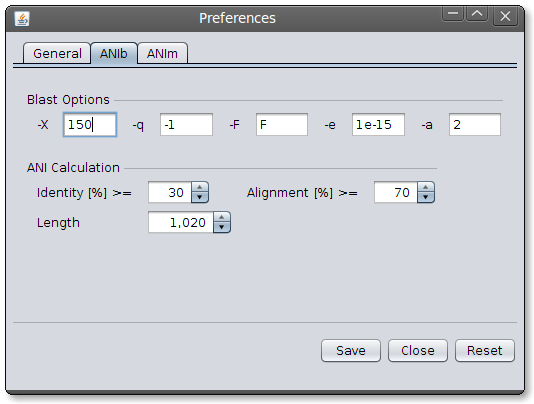 Image: JSpecies ANIb Parameters Dialog
Image: JSpecies ANIb Parameters DialogANIm parameters: JSpecies uses by default NUCmers default parameters. These parameters perform very well between close organisms (until around 80% of nucleotides aligned).
Warning: Always check the percentage of aligned nucleotides to weigh the reliability of ANIm values!
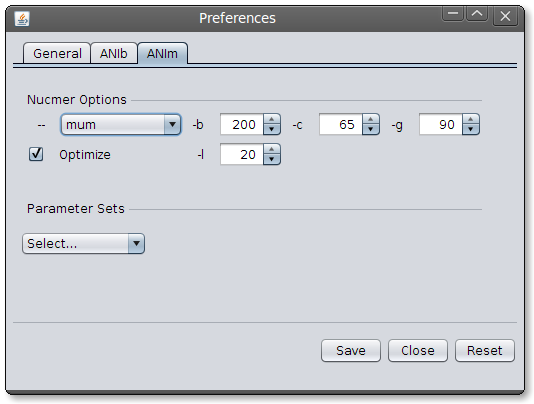 Image: JSpecies ANIm Parameters Dialog.
Image: JSpecies ANIm Parameters Dialog.3. Creating a new Group
Once the program has started, first the files to be compared must be downloaded. If it is the first calculation with the files, those have to be stored as a group. By selecting the NEW button, one can save the session name. The session can be reopened from the OPEN GROUP button if they have been previously stored. No additional calculations will be necessary.
Upon created the group name, files should be downloaded either from a fasta file in the personal directory [Import FASTA(S) from File(s)], or directly from the NCBI website (www.ncbi.nlm.nih.gov/genomes/lproks.cgi) [Import FASTA(S) from WWW].
 Image: JSpecies after initial start.
Image: JSpecies after initial start.4. Importing
Selecting the files from NCBI database: the interface should be connected with the remote database by pressing the CONNECT button. If the open session had previously been connected, just push the RELOAD button. Select the genomes to be downloaded, and press DOWNLOAD. The process of obtaining the files will take some minutes depending on the connexion, number of files and size.
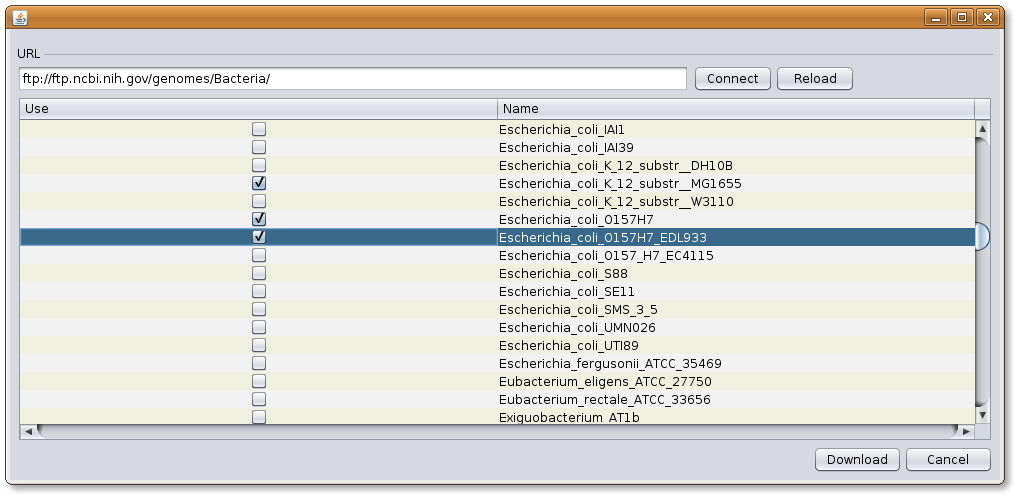 Image: JSpecies FTP download dialog.
Image: JSpecies FTP download dialog.Downloaded files: once the files have been completely downloaded, several menus will be shown. In the upper part the different files will be shown in different lashes. By selecting the desired file, the information on the sequence will appear in the window below. The graphic will shown the number of fragments (genomes and plasmids) that will be taken into account with its size.
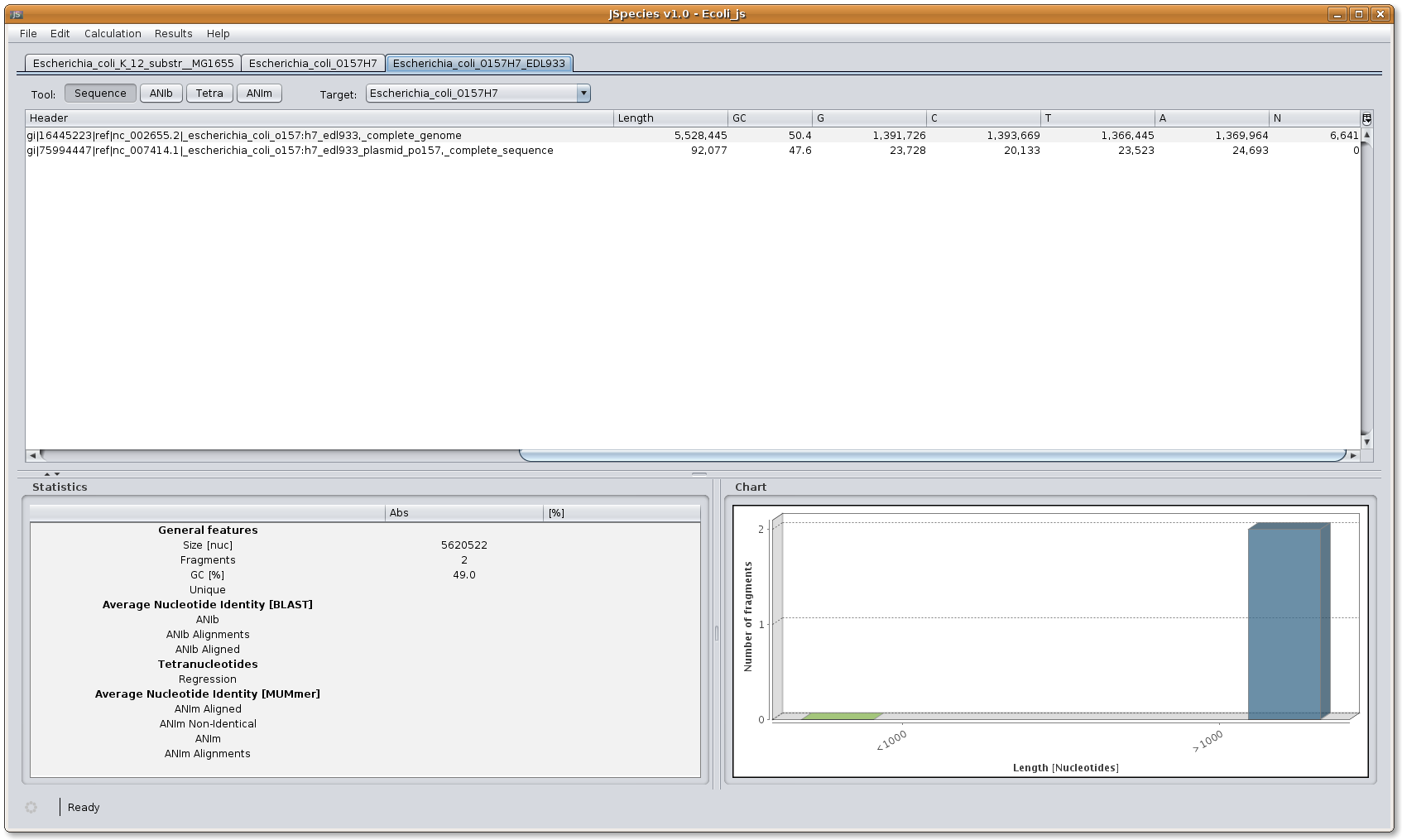 Image: JSpecies ready to start calculations.
Image: JSpecies ready to start calculations.5. Calculation
Starting the calculation of the different parameters: JSpecies can calculate ANIb (based on BLAST algorithm), ANIm (based on MUMmer algorithm) and TETRA. By pressing the button CALCULATION in the top menu, a new window will pop out in where one can select the different pair of files to be compared and the different parameters to be calculated. Upon selection, press the START button. Calculation may take minutes to hours depending on the number of files to be calculated. ANIb may take longer as it is a slow algorithm.
 Image: JSpecies calculation selecion dialog.
Image: JSpecies calculation selecion dialog.6. Navigating
Selecting the general features button: the menu allows the selection of the following results: Sequence (shows the features of the fasta files downloaded, i.e. number of chromosomes and plasmids with its length, GC and each nucleotide amount); ANIb (results of the BLAST comparisons); TETRA (results of the regression of the signature occurrences); ANIm (results of the MUMmer comparisons).
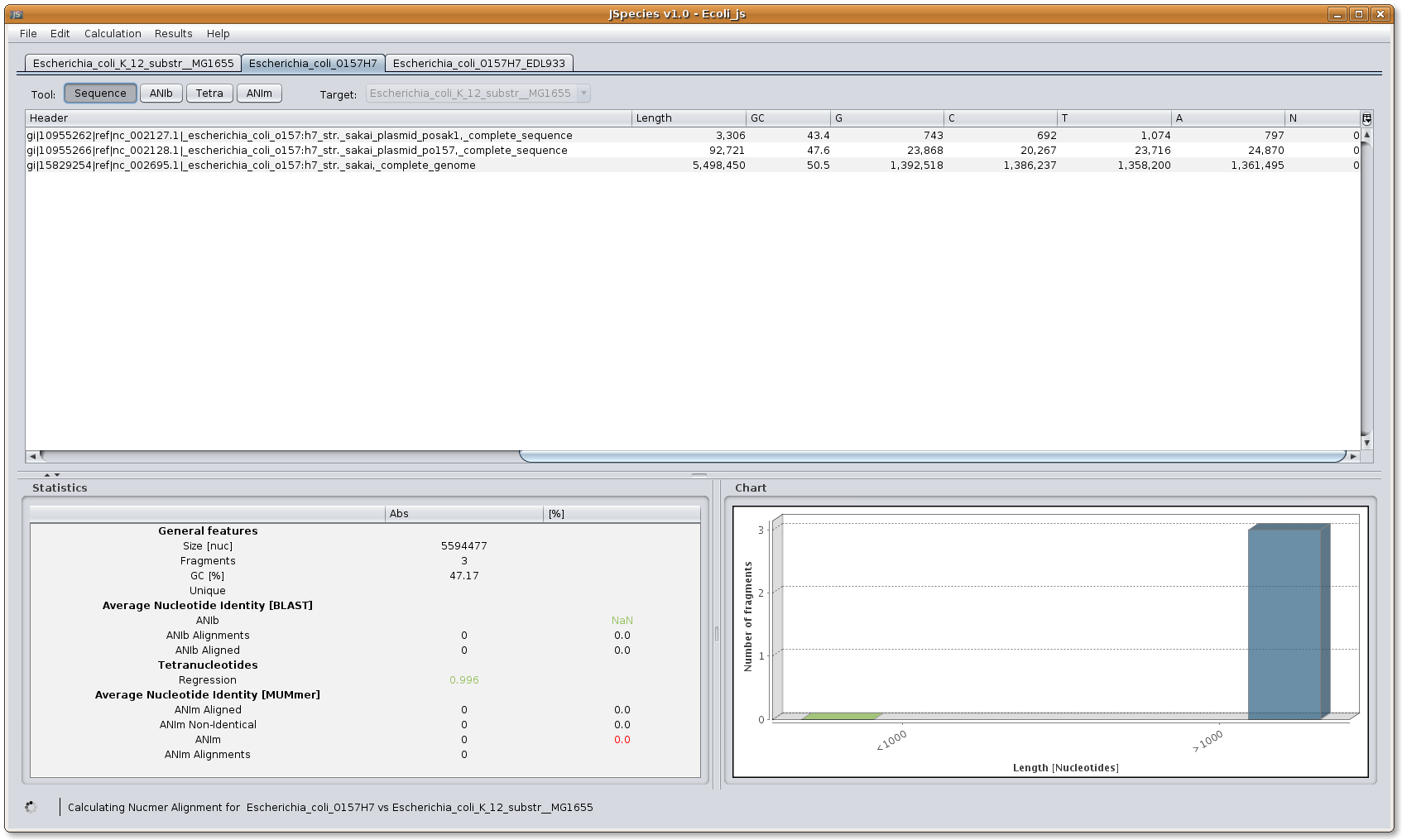 Image: JSpecies select a tool.
Image: JSpecies select a tool.Selecting strain lash: the menu allows the selection of the query fasta file being compared.
 Image: JSpecies select a organism.
Image: JSpecies select a organism.7. Overview ANIm (based on MUMmer)
ANIm menu: the menu shows the different fragments of the query fasta file used for calculation with its different lengths and position within the file. The chart window shows the distribution and number of fragments used to calculate ANIm. The statistics window shows the overall results calculated. ANIm aligned indicates the number of nucleotides used for calculation and its percentage corresponding to the complete query fasta file. ANIm Non-Identical shows the number of nucleotides not used for calculation and the percentage corresponding to the complete query fasta file. ANIm shows the total nucleotides used to calculate the parameter, and the ANIm value. Values above 96% will be shown in green, and below 96% in red. ANIm alignments indicate the number of fragments used for calculation independently of their size.
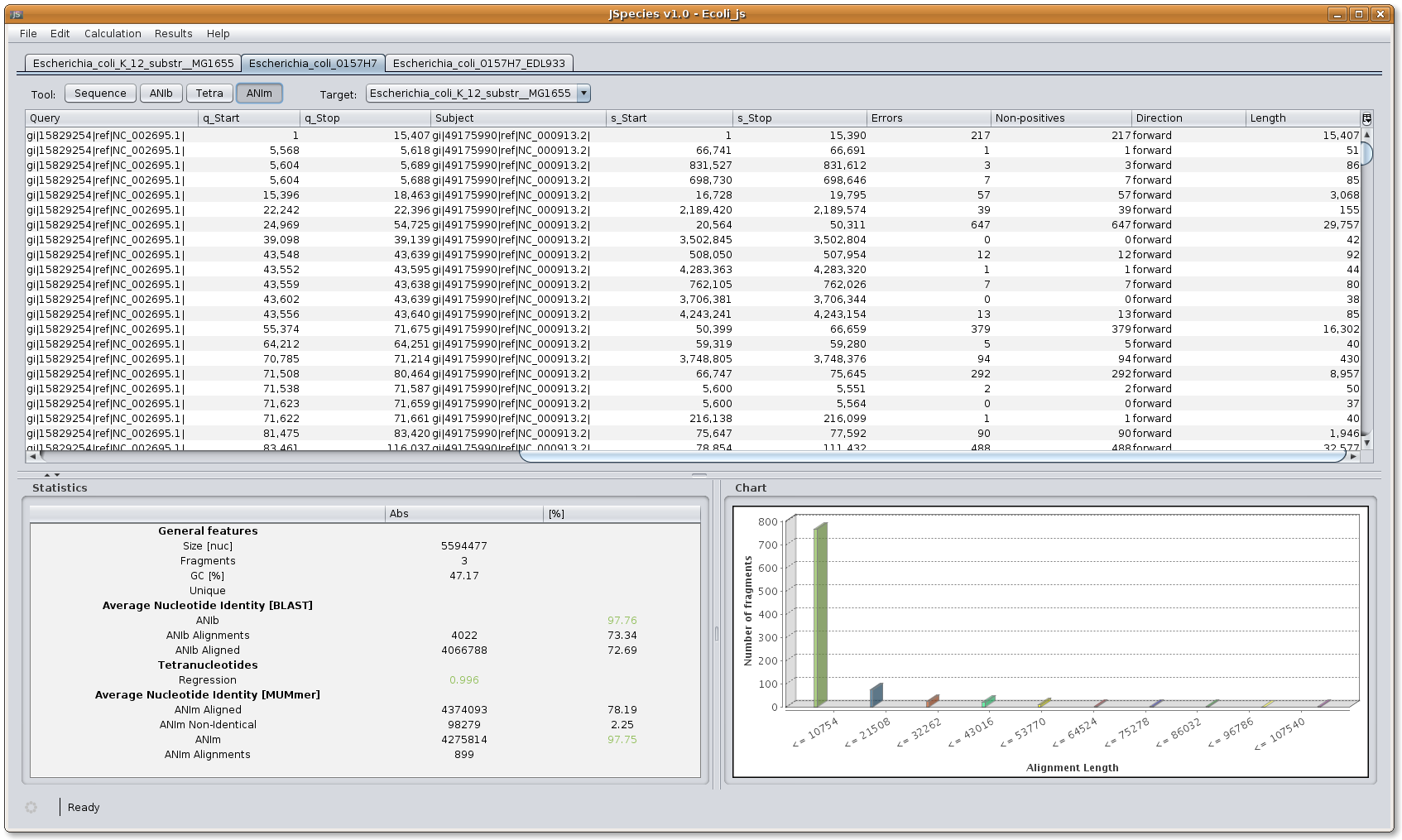 Image: JSpecies ANIm results overview.
Image: JSpecies ANIm results overview.8. Overview ANIb (based on BLAST)
ANIb menu: the ANIb menu shows the different fragments generated (default size of 1020 nucleotides), and the different fragments compared with their length and position in the fasta file. The chart shows the number of fragments compared distributed in the percentages of ANI identities. The statistics window shows the overall results calculated. ANIb will appear in green when the results are above 96% identity and in red if they are below 96% identity. ANIb alignments indicate the number of fragments used for comparisons, and the percentage corresponding to the total fragments generated. ANIb aligned indicated the total number of nucleotides compared and the percentage corresponding to the total nucleotides present in the query fasta file.
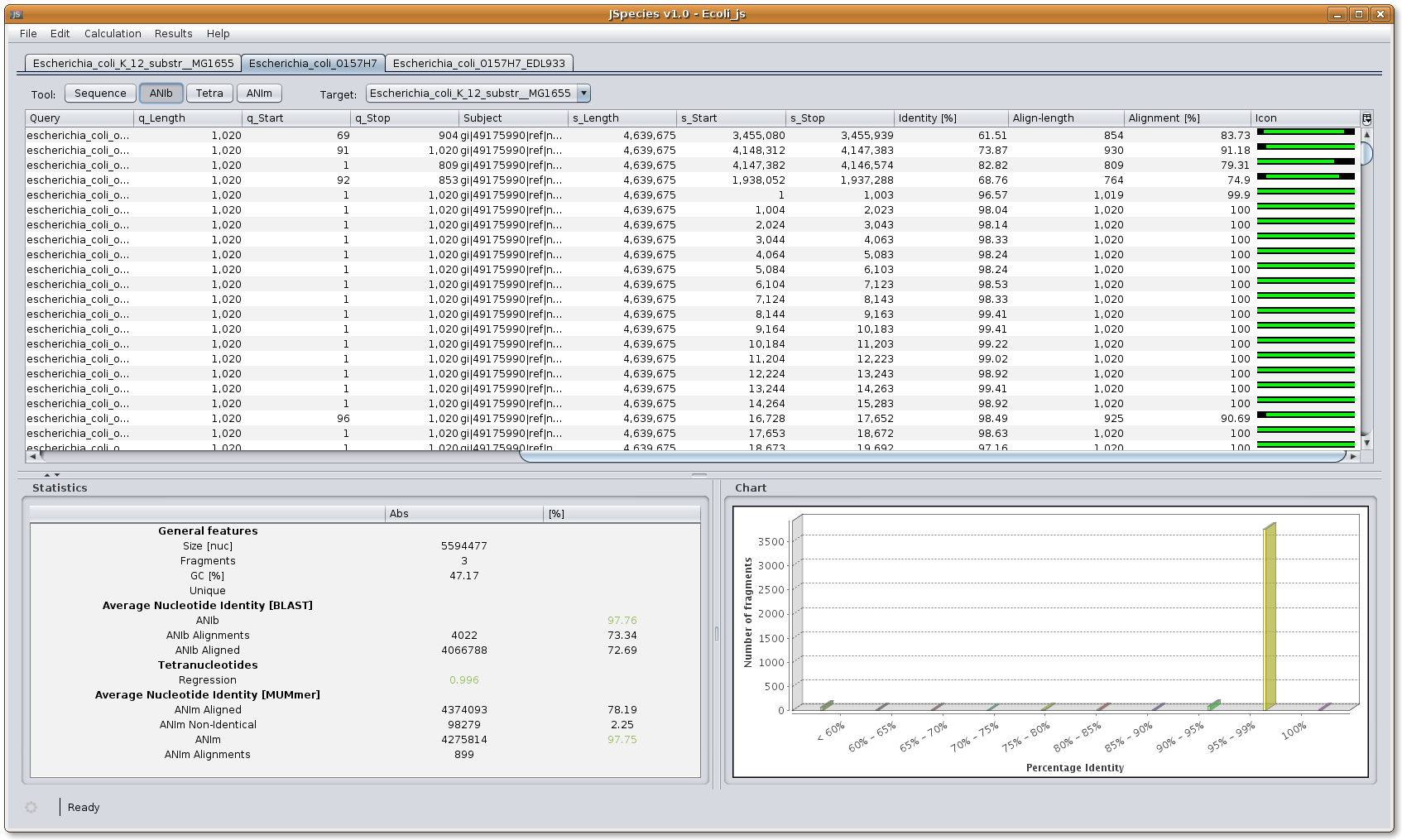 Image: JSpecies ANIb results overview.
Image: JSpecies ANIb results overview.9. Overview Tetra
TETRA menu: the menu shows the occurrence of each single tetranucleotide signature in the query fasta file and in the target fasta file. The chart shows the signature distribution plotting the query and target fasta files. The statistics window shows the regression coefficient resulting from plotting both signature occurrences.
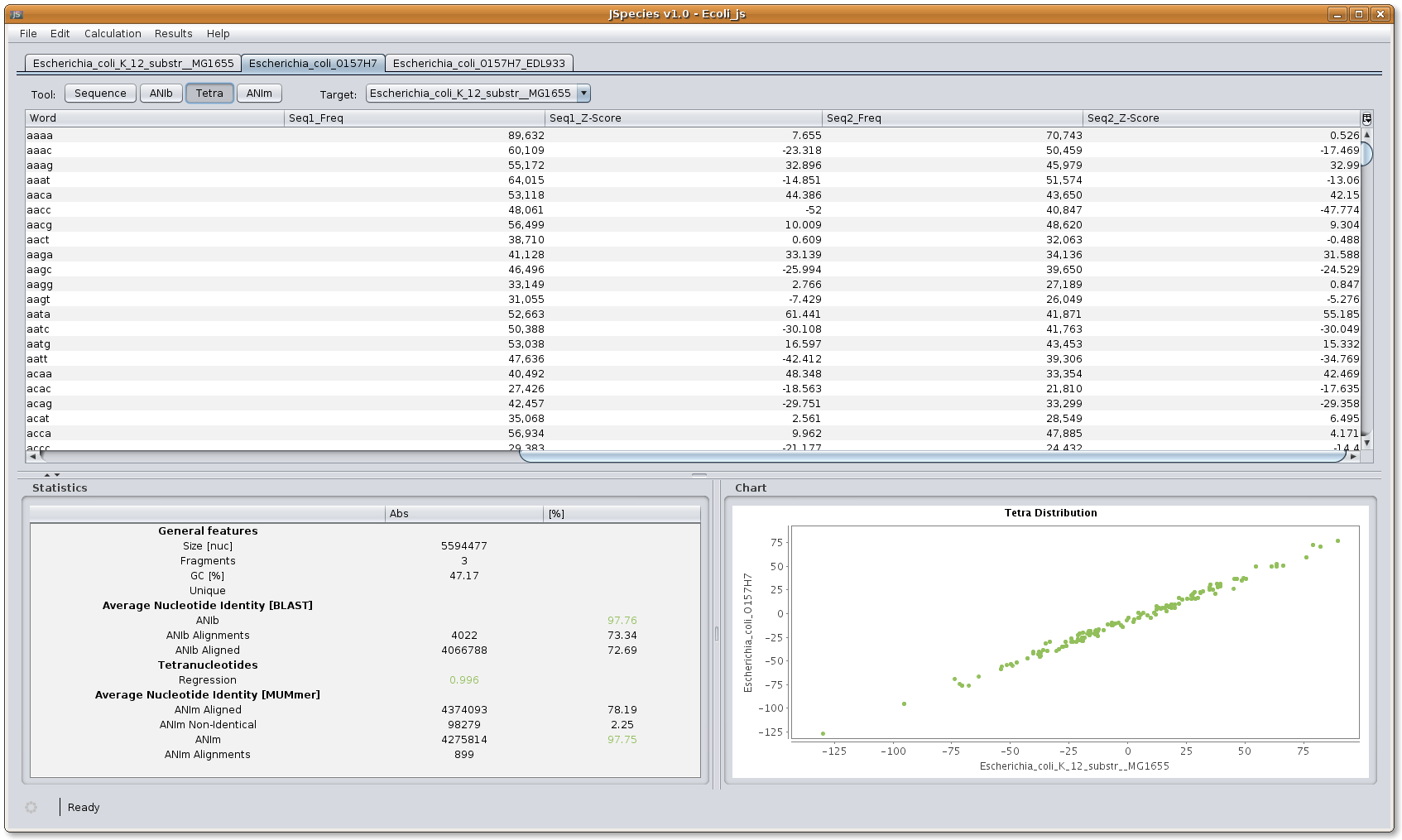 Image: JSpecies Tetra results overview.
Image: JSpecies Tetra results overview.10. Results: Show
Results menu: pressing the RESULTS button in the top menu a new window will pop out with the results calculated. One can select the different parameters that will be shown as an output matrix. The matrix can be copied and exported to an excel or word file.
 Image: JSpecies groups results overview.
Image: JSpecies groups results overview.